INTRODUCTION
Oncology is the study of disease caused by a tumor in the general sense that the tumor is a lump or abnormal pembekakan in the body. In terms of specific tumor is a lump caused by neoplasms. In general, tumors can be caused by neoplasm and nonneoplasma.
Neoplasms can be differentiated malignant and benign. Malignant neoplasms or cancers occur because of the rise and proliferation of cells in an uncontrolled manner so that these cells continue to grow and deform the growth of organ function. Cancer, carcinoma and sarcoma infiltrating grow (infultratif) into the surrounding tissue while destroying (destructive), can spread to other parts of the body and is generally fatal if left unchecked. Benign neoplasms grow with firm boundaries and does not infiltrate, but growing and pressing the tissue around it (expansive) and generally do not metastasize. Pathologic tumor classification based on the results of microscopic examination of tissue and tumor cells.
Tumor cells are the body's cells are transformed and grow out of control autonomic normal cell growth so that these cells differ from normal cells in the form and structure. Differences in the nature of tumor cells depends on the magnitude of the deviation in form and function, autonominya in growth, and ability to conduct infiltration and cause metastasis.
Tumor cells many forms (polimorfi) with multiple colors (polikromasi) due to high levels of nucleic acids in the core of the uneven distribution of chromatin core. Relatively large nucleus with the nucleus or cytoplasm ratio is lower. Higher incidence of mitosis and mitosis are abnormal. Irregular arrangement of cells (anaplastic). Tumor cells are grown on without limit so that the longer the larger the tumor and malignant neoplasms urgent sekitarnya.Pada tissue, the cells grow while infiltrate and seep into the surrounding tissue.
In many ways, childhood cancer is different from adult cancers. Childhood cancer is only about 2 percent of all cancers in humans, but it gives a disparate impact and is a significant cause of death in children. Generally, childhood cancer is not easy to know at an early, because at this early stage, rarely giving the patient complaints, and symptoms are easily seen.
Until now the cause of cancer in children is still not completely clear. But is believed to be due to the interaction of various factors (multifactorial). One suspected cause is the deviation of cell growth due to defective genes. Children do not have long enough exposure, so that genetic factors also play a role. Due to defects in the cell, cell growth so distorted and no longer want to interact with the environment dikendalikan.Pengaruh (ekogenetik factor) and various congenital defects or abnormalities are also suspected to participate increases the risk of childhood cancer. Another possibility is a factor of preconceptions. This can occur in mothers who have high risk, such as working in the area of radiation signal.
Symptoms of abdominal tumors in children and infants known to be more difficult, because children can not feel or tell his complaint. Therefore, the role of the people around him, especially the elderly, it is important to recognize the symptoms of brain tumor in children, so it can be immediately consulted the doctor.
Abdominal tumors in infants and children the most is the Wilm tumor (80%), and hepatoblastoma (65%). The tumor is derived not from intra-abdominal but most often manifests in the abdomen adalah Neuroblastoma, a number of events reached (70%).
Wilm tumor is a malignant tumor derived from embryonal renal blastema proliferation patologim metanefron due to the absence of normal stimulation of the duct metanefron tubule and glomeruli to produce a differentiated good. This tumor was found on clinical symptoms such as hypertension and hematuria. Diagnosis of these tumors based on clinical findings, radiological and histopathological examination.
Hepatoblastoma is a malignant liver tumor in children who are the cause remains unclear. This tumor was found on clinical symptoms of an enlarged abdomen, right abdominal pain, yellow and anorexia. Diagnosis of these tumors based on anamnesis, physical examination, laboratory and image radiologiknya.
Other abdominal solid tumor that most often is neuroblastoma. Krista neuroblastoma cells derived from neural sympathetic nervous system that manifests in the abdomen. Clinical symptoms are not specific to the tumor, such as anorexia, fatigue and bone pain. In the diagnosis of neuroblastoma can with investigations such as ultrasound, CT scan or MRI of the chest and abdomen.
The third type of tumor, have a course of treatment varies depending on patient age, tumor size and spread. Therapy may include removal of the tumor mass (surgery), chemotherapy and radiology.
To find out more about the most common tumor in children that manifests in the abdomen in the next chapter will discuss the definition, incidence, etiology, classification, pathology, clinical manifestations, diagnosis, treatment, prognosis and conclusions regarding each type of abdominal tumor.
Definition
Intra-abdominal tumor was the growth of a network with the multiplication of cells is uncontrolled and progressive that manifests in the abdomen which is characterized by the presence of a solid mass that is not pain and other symptoms depending on its place of origin of these tumors arise.
Incidence
The annual incidence of tumors in children less than 15 years an estimated 14: 100,000 population for the years 1986-1987. Malignancy remains a major cause of death from the disease between the ages of 1 and 15 years because there are approximately 6500 new cancer cases each year in this age group.
Etiology
Cause of the tumor is due to the occurrence of an abnormal cell division. Differences in the nature of tumor cells depends on the magnitude of the deviation in form and function autonominya in growth, ability to conduct infiltration and cause metastasis. There are several factors that can cause tumors include: carcinogens, hormones, lifestyle factors, parasites, genetic, infection, trauma, hipersensivitas.
According to the above incidence of intra-abdominal tumors in children are common are:
Wilm tumor
Wilms Tumor (Nephroblastoma) is defined as a kidney tumor that grows from primitive embryonic kidney cells. Wilms tumor is a malignant tumor derived from embryonal kidney metanefros .. Wilms tumor is a malignant kidney tumor that most infants and children. Approximately 80% of these tumors occur in children under 6 years, with a peak incidence at age 2-4 years. Wilms tumor can also be found in neonates. Wilms tumor accounting for 6% of all malignant disease in children. Incidence of disease is almost the same in every country, because there is no distinction of race, climate and environment, that is an estimated 8 per 1 million children under the age of 15. Comparison of the incidence of men and women are almost equal. Location of the tumor is usually unilateral, more often on the left, could also be bilateral (approximately 5%).

Figure 1. Wilm's tumor
Wilms tumors derived from pathological proliferation of blastema metanefron due to the absence of normal stimulation of the duct metanefron tubule and glomeruli to produce a differentiated good. Development of renal blastema to form kidney structures occur at 8-34 weeks gestation. So that the primitive blastema is estimated that the ability to pave the way towards the formation of Wilms tumor, whether as a germinal or somatic mutasi, it occurs at the age of 8-34 weeks gestation.
Approximately 1.5% of patients had a sibling or other family members who are also suffering from Wilms tumor. Almost all cases of unilateral descent is not different to the case of bilateral tumors. Approximately 7-10% of cases of Wilms' tumor inherited autosomal dominant. Genetic mechanisms associated with this disease, is not fully known. In patients with WAGR syndrome (Wilms tumor, aniridia, genital malformations and mental retadasi) shows the presence of cytogenetic deletions on chromosome 11, region p13. In some patients, found WT1 gene on the short arm of chromosome 11, region p13. WT1 gene expression specifically in the kidney and is known as transcription factors that are allegedly responsible for the development of Wilms tumor.
Pathology
Wilms tumor is composed of a network of primitive blastema metanefrik. Besides, these tumors often contain tissues that are not usually found in normal metanefron, such as bone tissue, cartilage and squamous epithelium. Histological picture of a very diverse is a hallmark of Wilms tumor. Classical picture is triphasic Wilms tumor, including stromal cells and epithelial blastema. Based on the correlation of histologic and clinical, histopathologic picture of Wilms tumor can be classified into three groups, namely low-risk tumors (favorable), tumor and tumor risk was high risk (unfavourable).
Stadium
The National Wilms Tumor Study (NWTS) dividing the 5-stage Wilms tumor, namely:
Stage I: Tumor confined to the kidney tissue without penetrating the capsule. This tumor resection can be completed.
Stage II: Tumor extends through the capsule and into the tissue surrounding the kidney and perirenal tissues, namely kidney, the renal hilum, renal vein and para-aortal lymph nodes. Still able to resect the tumor completely.
Stage III: Tumor spread to the abdominal cavity (perkontinuitatum), for example the liver, peritoneum, and others.
Stage IV: Tumor hematogenous spread to the abdominal cavity, lungs, brain and bones.
Clinical manifestations
Hematuri (macroscopic) present in approximately 25% of cases, due to tumor infiltration into the Calix system. Hypertension was found in about 60% of cases, presumably due to suppression of tumor or a hematoma in the blood vessels that supply blood to the kidneys, resulting in tissue ischemia which would stimulate the release of renin, or renin release tumor itself. Other symptoms of anemia, weight loss, urinary tract infection, fever, malaise and anorexia. In some patients could be found who are colicky abdominal pain, due to blood clots in the urinary tract. Wilms tumor is rarely found with other congenital abnormalities, such as aniridia, hemihipertrofi, urinary tract or genital anomalies and mental retardation.
Diagnosis
Wilms tumor diagnosis based on:
• Clinical symptoms
• radiological examination (IVP and ultrasound), laboratory LDH
• Ensure the histopathological examination of tumor tissue
With visible distortion IVP examination pielokalises system (changes in shape pielokalises system) and at the same examination is useful to determine kidney function. Ultrasound is a noninvasive examination that can differentiate solid tumor with tumor-containing fluid. With ultrasound, Wilms tumor appears as a solid tumor in the kidney area. Laboratory findings are important for supporting the Wilms tumor is the degree lactic dehydrogenase (LDH) rises and Vinyl mandelic acid (VMA) within normal limits.
Therapy
Modality treatment consisting of Wilms tumor, surgery (surgery), chemotherapy and radiotherapy. On stage I and II tumors with favorable cell type, performed the operation with dactinomycin and vincristine combination chemotherapy without abdominal radiation delivery. Stage III tumors with favorable given cell type by a combination of surgical treatment daktinomisin, vincristine and doxorubicin with abdominal radiation. For stage IV tumors with favorable cell types, given the combination daktinomisin, vincristine and doxorubicin. This patient also had abdominal radiation and lung cancer when it is no spread to the lung tissue. In the case of stage II to IV with a type of cell anaplastic (unfavorable) are given surgical treatment with a combination daktinomisin, vincristine and doxorubicin plus siklofospamid. On these patients also received radiation abdomen and lungs.
Prognosis
Several factors determine the prognosis, the tumor size, histopathologic picture, patient age and tumor stage or rate of spread. Those who have a good prognosis is the patient who had a tumor size was small, high-level histopathologic differentiation of cells, is still an early stage or metastasis, and no person under the age of two years.
Hepatoblastoma
Liver tumors in children can be benign or malignant (cancer), and can be a primary or a metastasis from another organ. Rare primary liver tumor in children, there are approximately 3% of all tumors in children. Approximately 50-60% of liver tumors in children are malignant and more than 65% of whom were hepatoblastoma. Other malignant tumors that are often in getting the child is a hepatocellular carcinoma.
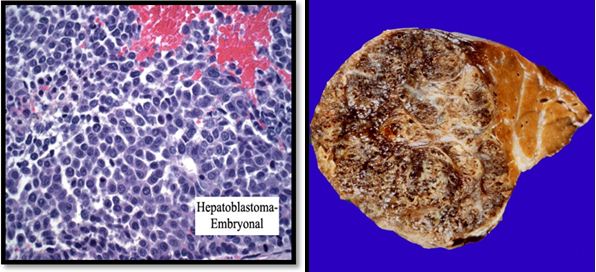
Figure 2. Picture as embryonal hepatoblastoma cells and anatomic pathology
Pathology
Causes of liver tumors is still unclear. Tumors were found at birth are generally benign, such as hamartoma, hemangioma, and hemangioendotelioma. There are 2 types of malignant tumors are often found in children are hepatoblastoma and hepatocellular carcinoma. Hepatoblastoma are found under 3 years old and was associated with a genetic disorder that is loss of heterozygosity on chromosome 11p15 that cause interference with the tumor suppression gene. Hepatoblastoma is also commonly associated with the Beckwith-Weidemann syndrome where there is a disruption in the gene insulin-like growth factor-II with the clinical manifestation of exophthalmos, gigantism, macroglossia, microcephaly, and viceromegaly. Hepatocellular carcinoma in children can be found from birth to age 19 years, usually associated with hepatitis B or C infection, chronic tirosinemia hereditary type, glycogen storage disease, α1-antitrypsin deficiency and biliary cirrhosis.
Clinical manifestations
Some symptoms are often available include:
• The large abdomen, or distention
• Abdominal pain right
• decreased appetite, weight loss
• Vomiting
• Jaundice
• Heat
• Itching of the skin
• Anemia
• Back pain due to tumor suppression
• It can also happen with the outbreak of the crisis of acute abdominal tumor and hemoperitonium (usually in hepatocellular carcinoma).
Diagnosis
Besides anamnesis and complete physical examination, several investigations are also required in establishing the diagnosis of liver tumors in children and metastasenya, including:
• Lab: CBC, blood chemistry, liver and kidney function tests, hepatitis B and C serology; α-fetoprotein/AFP (also for monitoring therapy) Liver biopsy for histopathological examination
• Radiology: Plain chest, ultrasound / Doppler ultrasound, CT-scan/MRI
There are several methods of staging liver tumors in children, one of them as follows:
• Stage I:
complete tumor can be removed by surgery
• Stage II:
tumor can be removed surgically but still leave some residual
• Stage III:
tumor can not be removed completely with surgery and found the spread in lymph nodes surrounding
• Stage IV:
tumor has spread to other organs.
Therapy
Treatment based on type and stage of tumor:
• hepatoblastoma stage I and II: tumor removal and chemotherapy followed by 4 series using cisplatin, vincristine, and fluorouracil.
• hepatocellular carcinoma stage I and II: tumor removal followed by chemotherapy or cisplatin and doxorubicin.
• stage III and IV hepatoblastoma: Some alternative treatments can be done:
A. Chemotherapy to reduce tumor size followed removal of as much as possible of the tumor and chemotherapy is closed again.
2. Tumor metastases in lung surgery.
3. Chemotherapy.
4. Radiotherapy followed by surgery.
5. Injecting chemotherapy drugs directly into the liver vein.
6. Chemotherapy and kemoembolisasi.
7. Liver transplantation.
• hepatocellular carcinoma stage III and IV: The reduction of tumor size using cisplatin chemotherapy with vincristine / fluorouracil or doxorubicin followed removal of the tumor as possible.
• Do re-treatment relapse based on prior treatment.
Prognosis
Prognosis of malignant liver tumors varied in each patient. Rapid and precise diagnosis and treatment of progressive essential to get the best prognosis for patients. Children with benign liver tumors usually will not have problems later on after the tumor is removed. Still need to develop new treatment methods that can enhance the therapeutic efficacy and side effects lowest possible pressure.
Neuroblastoma
Neuroblastoma is a childhood extracranial solid tumor that most frequently, covering 8-10% of all childhood cancers, and an infant neoplasms are diagnosed most frequently. The median age at diagnosis is 2 years; 90% were diagnosed before 5 years. The annual incidence 8.7 per million children, or 500-600 new cases each year in the United States. The incidence was higher in men and in whites. Microscopic group neuroblasts normally found in the fetal adrenal gland and approximately 1 in 200 neonates at the time of autopsy.

Figure 3. Manifestations of abdominal neuroblastoma in infants aged 18 months.
Pathology
Krista neuroblastoma cells derived from neural sympathetic nervous system and because it can arise anywhere from the posterior cranial fossa through koksik. Approximately 70% of these tumors arise in the abdomen, 50% of that amount in the adrenal gland 20% arise in the thorax, usually in the posterior mediastinum. Tumors were most often spread to surrounding tissues with local invasion and regional lymph nodes via the lymph nodes. Haematogenous spread to the bone marrow, skeleton and liver are common. With immunological techniques can be detected in tumor cells in peripheral blood of more than 50% of children at time of diagnosis or relapse. The spread to the brain and lungs occurs in rare cases.
The clinical manifestations
The initial symptoms are usually nonspecific, such as loss of appetite, fatigue, and bone pain. The symptoms usually associated with the origin and spread of tumor:
• derived from stomach cancer usually symptoms of an enlarged abdomen, the stomach feels full, abdominal pain, difficult bowel and bladder, and blood pressure may also increase.
• Cancer that has spread to the bone will cause bone pain
• Cancer that has spread to the bone marrow leads to:
- Reduced number of red blood cells resulting in anemia
- Reduced number of platelets so that children prone to bruising
- Reduced number of white blood cells so that the child vulnerable to infection.
Approximately 90% of neuroblastoma produces hormones (eg epinephrine, which can cause increased heart rate and occurrence of anxiety).
Other symptoms that may be found: pale skin, the dark circles around the eyes appear, chronic fatigue, excessive tiredness lasting for weeks or months, diarrhea, feeling unwell (malaise) lasting for weeks or months , excessive sweating, uncontrollable eye movements and fussy.

Diagnosis
If the cancer grows large enough, may be palpable on abdominal examination. If the tumor has spread to the liver, enlarged liver palpable. Adrenal gland tumors often cause high blood pressure and rapid heartbeat.
Other checks are wont to do:
• abdominal ultrasound and chest
• CT scan or MRI of the chest and abdomen
• Chest X-rays
• Biopsy of the liver, lungs, skin, bone marrow
• Bone Scan
• Complete blood.
• Examination of urine to see the formation of excessive hormone epinephrine.
Therapy
Treatment varies, depending on patient age, location, size and spread of cancer has not spread tumor.Bila, usually removed surgically. If the cancer is large or has spread, chemotherapy is given (anti-cancer drugs vincristine, cyclophosphamide, doxorubicin and cisplatin) or radiation therapy. If the cancer has spread to several organs or high risk, usually given external radiotherapy.
Prognosis
Prognosis depends on the child's age, tumor size and extent of tumor spread. In children younger than 1 year and the tumor is small and has not spread, the prognosis is very good. Some patients experience spontaneous regression, in which tumor tissue maturation and develop into benign ganglioneuroma, which can be removed surgically. In other cases, the tumors spread quickly to other organs. Response to treatment varies, if the treatment is performed before the tumor spreads, then the result is usually satisfactory.
RHABDOMYOSARKOMA
Epidemiology and Etiology
Rhabdomyosarkoma is the most common type found SJLA, ie +60% in SJLA under 5 years and +23% in children 15-20th, and found slightly higher in boys.
Adalah multifactorial etiology factors and the role of familial factors have been studied for its role in children often dihungkan rhabdomyosarkoma with Li-Fraumeni syndrome, Beckwith-Weidsmann syndrome and Neurofibromatosis-1 (NF-1).
Common is the location of the orbit and intraabdominal, genitourinary. Besides, it can also occur intratorakal and lower extremities.
Type Histopathology
Rhabdomyosarkoma in children divided into:
- Embryonal rhabdomyosarcoma
- Alveolar rhabdomyosarkoma
- Spindle cell rhabdomyosarcoma
- Botryoid rhabdomyosarcoma
- Undifferentiated rhabdomyosarcoma
- Rhabdomyosarcoma with rhabdoid features
Stadium Clinic
Based on TNM staging preterapi
Diagnostic Procedures
Based on anamnesis and physical examination in accordance with the most common location rhabdomyosarkoma children, including the examination of regional lymph nodes and distant metastases. Tumor location in retrobulbair may be proptosis or bumps. And at other locations in the form of a lump with normal overlying skin, may no complaints or accompanied by pain. Investigations include plain or CT-scan in the primary tumor and metastases at distant places. If you need to do well in the bone marrow aspiration biopsy. Definitive diagnosis is from the biopsy incision / excision.
Therapeutic Procedures
Depending on the location of the primary tumor and is associated with histopathological types and are encouraged to perform a multimodality and multidisciplinary therapy, it is not advisable to conduct an aggressive mutilation.
A. Orbital and parameningeal locations including the middle ear and nasopharynx. Performed up to 5000 cGy of radiotherapy or chemotherapy with a combination of vincristine, Dactinomycin and Doxorubicin.
2. Locations in the non-orbital non-parameningeal and covers the region of the parotid, larynx, palate, tonsil, glosis / tongue, buccal / cheek, nasal / nose, head and neck. Where possible should be done excision followed by adjuvant radiotherapy to the 4500-5000cGy or chemotherapy given vincristine, Dactinomycin and Cyclophosphamide (VAC).
3. Location in the thoracic wall, intrathoraks, abdominal wall, paraspinal and retroperitoneal. The main therapy is radical excision, if necessary, be given adjuvant radiotherapy when the embryonal type.
4. Locations in the extremities. Untujk radical excision is recommended to limit the freedom of a microscopic incision. Not recommended for action compartment excision or amputation or excision of muscle groups. If need be given adjuvant radiotherapy to 5000cGy. Chemotherapy is not recommended due to poor response.
5. Locations in the genito-urinary. Radical resection whenever possible, if not impossible limited resection followed by adjuvant radiotherapy. If not possible resection, preoperative radiotherapy can be done or neoadjuvant chemotherapy with vincristine + Dactinomycin followed by resection
CONCLUSION
• In terms of specific tumor is a lump caused by neoplasms. In general, tumors can be caused by neoplasm and nonneoplasma.Neoplasma indistinguishable malignant and benign.
• Malignant neoplasms or cancers occur because of the rise and proliferation of cells in an uncontrolled manner so that these cells continue to grow and deform the growth of organ function.
• Cancer child is only about 2 percent of all cancers in humans, but it gives a disparate impact and is a significant cause of death in children. Symptoms of abdominal tumors in children and infants known to be more difficult, because children can not feel or tell his complaint.
• Abdominal tumors in infants and children who most are Wilm tumor (80%), hepatoblastoma (65%) and tumors derived not from intra-abdominal but most often manifests in the abdomen adalah Neuroblastoma, a number of events reached (70%).
• Wilm tumor is a malignant tumor derived from embryonal renal blastema proliferation metanefron pathology due to the absence of normal stimulation of the duct metanefron tubule and glomeruli to produce a differentiated good. This tumor was found on clinical symptoms such as hypertension and hematuria. Diagnosis of these tumors based on clinical findings, radiological and histopathological examination.
• hepatoblastoma liver are tumors that cause malignant in children remains unclear. This tumor was found on clinical symptoms of an enlarged abdomen, right abdominal pain, yellow and anorexia. Diagnosis of these tumors based on anamnesis, physical examination, laboratory and image radiologiknya.
• other abdominal solid tumor that most frequently is the neuroblastoma. Krista neuroblastoma cells derived from neural sympathetic nervous system that manifests in the abdomen. Clinical symptoms are not specific to the tumor, such as anorexia, fatigue and bone pain. In the diagnosis of neuroblastoma can with investigations such as ultrasound, CT scan or MRI of the chest and abdomen.
• Prognosis depends on tumor third to several factors, namely tumor size, histopathological picture, the age of the patient and the stage or degree of tumor spread.
REFERENCES
- Sjamsuhidrajat R, W. Neoplasia dalam Buku Ajar
Ilmu Bedah. Edisi Ke-2.
Jakarta :EGC. 2004; 7 : 131-135.
- Crist
Wiliam M. Penyakit Neoplasma dan Tumor dalam Behrman, Klegman dan Arvin
Ilmu Kesehatan Anak Nelson volume 3 edisi 15. Jakarta : EGC. 2000;445 :
1759
- http://ns-nining.blogspot.com/2008/10/tumor-abdomen.htm.
- http://ebookfkunsyiah.wordpress.com/2008/09116/Tumor padat pada ana
- Sharer
Patrics D, Yudith WA. Neoplasma Ginjal dalam Behrman, Klegman dan Arvin
Ilmu Kesehatan Anak Nelson volume 3 edisi 15. Jakarta : EGC. 2000;452 :
1784-86.
- Jacobson,
Dr. 2004. Hepatocellular Carcinoma. Last update : June 23, 2004. Available
at http://www.emedicine.com/radio/topic 332.htm.
- Santana
Victor M. Neuroblastoma dalam Behrman, Klegman dan Arvin Ilmu Kesehatan
Anak Nelson volume 3 edisi 15. Jakarta : EGC. 2000;451 : 1781-84.
- http://adulgopor.files.wordpress.com/2009/12/neuroblastoma.pdf.
- http://www.nant.org/patient
and famillies/neuroblastoma.php.
No comments:
Post a Comment